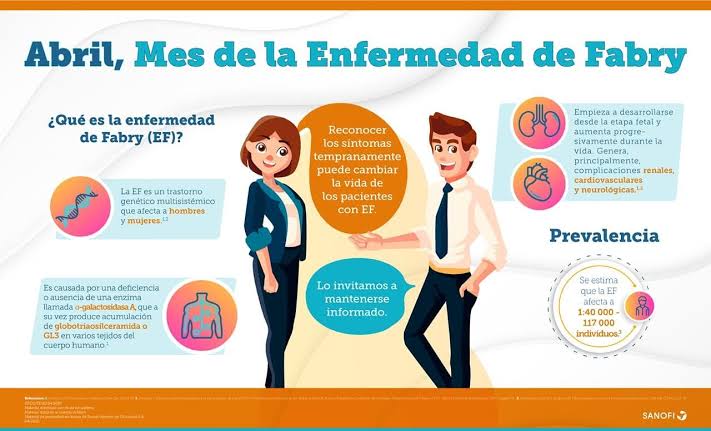
La enfermedad de Fabry es un trastorno genético raro, hereditario y multisistémico (ligado al cromosoma X) causado por la deficiencia de la enzima alfa- galactosidasa.
Esto provoca la acumulación de grasas (globotriaosilceramida – Gb3/GL-3) en las células, dañando órganos como corazón, riñones y sistema nervioso. Se manifiesta con dolor, angioqueratomas, hipohidrosis y complicaciones cardíacas/renales.
Características Principales
* Herencia: Ligada al cromosoma X. Afecta principalmente a hombres, mientras que las mujeres pueden presentar síntomas de intensidad variable.
* Acumulación Lisosomal: La falta de la enzima impide descomponer el GL-3, provocando depósitos progresivos en el endotelio vascular y otros tejidos.
Tipos:
Clásica: Inicio en la infancia o adolescencia con síntomas multisistémicos graves.
Tardía/Atípica: Aparición más tardía, a menudo afectando principalmente a un solo órgano (corazón o riñón).
Síntomas Comunes
Los síntomas suelen aparecer en la infancia y progresan con la edad:
Dolor neuropático: Dolor y ardor en manos y pies (acroparestesias), a menudo desencadenados por calor, frío o ejercicio.
* Piel: Angioqueratomas (pequeñas manchas rojas/moradas elevadas, frecuentemente entre el ombligo y las rodillas).
* Sudoración: Hipohidrosis (disminución de la capacidad de sudar) o anhidrosis.
* Ojos: Opacidades corneales (córnea verticilata), generalmente sin afectar la visión.
* Problemas gastrointestinales: Dolor abdominal, diarrea o náuseas.
* Afectación de órganos: Insuficiencia renal, hipertrofia ventricular izquierda (corazón) y riesgo de accidentes cerebrovasculares.

Diagnóstico y Tratamiento
Se confirma mediante análisis de sangre que miden la actividad enzimática
y pruebas genéticas para detectar la mutación.
El Tratamiento se centra en la Terapia de Sustitución Enzimática (TSE) para reemplazar la enzima deficiente. También se utilizan medicamentos para el dolor (gabapentina) y para proteger la función renal (inhibidores ACE